La bioinformática es un campo de la ciencia que combina la biología y la informática para analizar datos biológicos. Las herramientas bioinformáticas ayudan a organizar, comparar y analizar datos, predecir estructuras de proteínas, realizar análisis genómicos y apoyar la investigación.
En este artículo, he recopilado algunas herramientas que serán beneficiosas no sólo para los bioinformáticos, sino también para investigadores, científicos, analistas, estudiantes, etc.
Hablaré de varias herramientas que pueden ayudarle a analizar datos biológicos, realizar análisis estadísticos, visualizar datos, llamar variantes, predecir estructuras de proteínas, utilizar navegadores genómicos, realizar alineaciones de secuencias y mucho más.
OmicsBox
OmicsBox destaca como un software bioinformático de primera categoría, que proporciona un análisis exhaustivo de los datos de secuenciación de próxima generación (NGS) para genomas, transcriptomas y metagenomas. Es ampliamente utilizado por las principales instituciones de investigación a nivel mundial, diseñado para ser fácil de usar, eficiente y repleto de potentes herramientas para el manejo de grandes y complejos conjuntos de datos.
OmicsBox está estructurado en módulos, cada uno de ellos adaptado a análisis específicos, como el análisis genómico, la variación genética, la transcriptómica, el análisis funcional y la metagenómica.

Características principales
- El módulo de análisis genómico es un conjunto de herramientas eficiente y fácil de usar para caracterizar y analizar genomas recién secuenciados.
- El Módulo de Transcriptómica permite procesar los datos de ARN-Seq desde las lecturas en bruto hasta el análisis funcional de forma flexible e intuitiva.
- El Módulo de Análisis Funcional proporciona contexto biológico como opción de análisis.
- El Módulo de Metagenómica permite el análisis de los datos del microbioma, incluyendo el ensamblaje, la anotación y la clasificación de los datos metagenómicos.
OmicsBox proporciona un conjunto de herramientas para analizar datos genómicos, transcriptómicos y metagenómicos, lo que lo hace valioso para los profesionales que trabajan en biología computacional y bioinformática.
Bioconductor
Bioconductor, un esfuerzo de código abierto, incluye paquetes R especializados para interpretar datos genómicos de alto rendimiento. El lenguaje y entorno R para el cálculo estadístico y los gráficos ha sido mejorado por Bioconductor para hacer frente a los retos únicos que plantean la genómica y la bioinformática.
El completo conjunto de herramientas que proporciona Bioconductor permite a los usuarios realizar una serie de tareas, desde el preprocesamiento básico de datos hasta análisis estadísticos avanzados.
Características principales
- El conjunto de herramientas Bioconductor proporciona una amplia gama de paquetes R especializados para tareas como el análisis de datos de micromatrices, datos de secuenciación de alto rendimiento, datos de citometría de flujo, etc.
- Documentación de alta calidad e investigación reproducible
- Los paquetes Bioconductor suelen incluir funcionalidades para interactuar con bases de datos genómicas populares, como Ensembl y UCSC Genome Browser, y recuperar datos de ellas.
El conjunto de herramientas Bioconductor es ampliamente utilizado por bioinformáticos, investigadores y científicos que trabajan con datos genómicos de alto rendimiento.
FastQC
FastQC es una herramienta de control de calidad ampliamente utilizada y diseñada para datos de secuencias generados mediante métodos de secuenciación de nueva generación. FastQC ofrece un enfoque directo para el control de calidad de los datos brutos procedentes de los pipelines de secuenciación.
La herramienta se puede descargar fácilmente y cuenta con una interfaz fácil de usar, lo que permite a los usuarios abordar los problemas de los datos antes de proceder con el análisis posterior. Los archivos de entrada consisten en secuencias de lectura, y la herramienta genera la salida en forma de gráficos y resúmenes tabulares de los resultados.
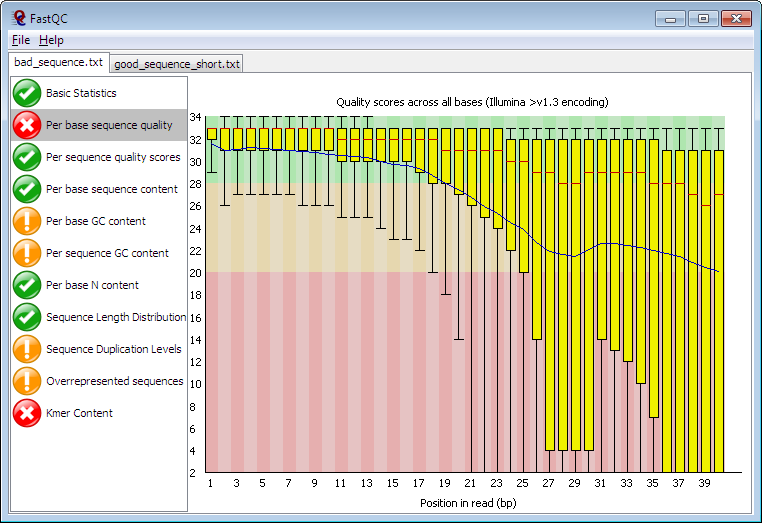
Características
- Los informes pueden generarse automáticamente sin necesidad de utilizar la aplicación interactiva cuando se está fuera de línea.
- Proporcionan una rápida visión general del problema.
- Importa datos de archivos BAM, SAM o FastQ (cualquier variante) para un cómodo análisis.
- Genera gráficos y tablas de resumen para la evaluación de la calidad de los datos
- Exportación de informes HTML
Es Ideal para procesar y analizar datos de secuenciación. Por lo tanto, es Recomendado para analistas de datos biológicos, investigadores y equipos de proyecto.
EMBOSS
EMBOSS (European Molecular Biology Open Software Suite) es una de las herramientas para aprender el análisis bioinformático si es usted principiante. EMBOSS es un paquete de software de vanguardia, gratuito y de código abierto, meticulosamente elaborado para satisfacer las demandas únicas de la comunidad de biología molecular y bioinformática.
EMBOSS tiene muchas herramientas para analizar secuencias, y funciona bien con otros paquetes y herramientas populares. Una de ellas es EMBOSS Needle:
Características principales
EMBOSS incluye más de 200 aplicaciones para:
- Mostrar características de una secuencia
- Búsqueda en bases de datos
- Presentación de datos de secuencias
- modelos moleculares en 3D
- Plataformas bioinformáticas
- División de una secuencia en secuencias más pequeñas
Es una herramienta fácil de usar para principiantes, útil para estudiantes, investigadores computacionales, bioinformáticos y científicos de datos.
Clustal
Clustal es un conjunto de herramientas bioinformáticas como ClustalX, ClustalW y Clustal Omega. Está diseñado para alinear secuencias biológicas como ADN, ARN y proteínas. Ayudan a identificar regiones similares en múltiples secuencias, ayudando en el análisis de las relaciones evolutivas y los dominios funcionales.
La serie de programas Clustal se utiliza ampliamente en biología molecular para la alineación múltiple de secuencias tanto de ácidos nucleicos como de proteínas y para la preparación de árboles filogenéticos.
Características principales
- Los programas Clustal suelen ofrecer interfaces fáciles de usar
- Clustal destaca en la alineación de tres o más secuencias
- Clustal X ayuda a predecir la alineación de secuencias múltiples y el análisis filogenético para secuencias genéticas dadas de diversos organismos.
- Clustal Omega utiliza una heurística basada en el análisis filogenético
Clustal es el más adecuado para los profesionales de los campos de la bioinformática y la biología computacional que necesitan alinear y analizar múltiples secuencias biológicas.
DNASTAR Lasergene
DNASTAR Lasergene es un paquete de software de vanguardia que sirve como solución versátil para la biología molecular, la genómica y el análisis de proteínas. Científicos de todo el mundo confían en esta completa plataforma.
Además, DNASTAR ofrece Nova Applications, que incluye NovaFold AI, NovaFold, NovaFold Antibody y NovaDock, mejorando aún más sus capacidades en la predicción y el análisis de la estructura de proteínas.

Características principales
- Soluciones integrales para biología molecular, anticuerpos, genómica y análisis de proteínas, que ofrecen una suite completa para los investigadores.
- Automatiza las tareas en proyectos de genómica, agilizando el ensamblaje y el análisis de secuencias.
- Proporciona representaciones gráficas 3D flexibles y ricas de las estructuras proteicas, mejorando la comprensión de las secuencias de proteínas.
DNASTAR Lasergene, con sus completos paquetes y sus avanzadas aplicaciones Nova, es una suite de software versátil que satisface las diversas necesidades de los biólogos moleculares, los investigadores en genómica, los científicos especializados en proteínas y los bioinformáticos estructurales.
GATK
El kit de herramientas para el análisis del genoma (GATK), creado por la plataforma de ciencia de datos del Instituto Broad, es una potente herramienta para el descubrimiento de variantes genéticas y el genotipado. Sobresale en el procesamiento de grandes archivos de entrada y se centra en la identificación de variaciones como SNP e indels en datos de ADN y ARN-Seq.
GATK también maneja números de copias y variaciones estructurales e incluye utilidades para el control de calidad en la secuenciación de alto rendimiento.

Características principales
- GATK está optimizado para obtener resultados precisos y de alta calidad.
- GATK emplea un marco de programación estructurado, aprovechando la filosofía de programación funcional de MapReduce.
- Maximiza la eficiencia computacional para un procesamiento eficaz.
- Capaz de realizar análisis genómicos para exomas y genomas completos.
- Ofrece los mejores flujos de trabajo para variantes cortas somáticas.
GATK es una herramienta para quienes pretenden navegar por las complejidades del análisis genómico con precisión y facilidad, como investigadores genómicos, científicos y bioinformáticos.
Software de análisis universal
El Software de Análisis Universal (UAS) simplifica el análisis y la gestión de los datos genómicos forenses. Es compatible con varios flujos de trabajo ForenSeq, procesa rápidamente los datos y proporciona llamadas de variantes fiables sin necesidad de licencias por puesto.
Adaptado a los analistas forenses, UAS agiliza el manejo de la información de secuencias, facilitando una revisión eficaz de los perfiles de ADN. UAS está diseñado para ser compatible con el sistema de secuenciación MiSeq FGx y con las herramientas habituales de terceros, lo que lo convierte en una solución versátil y fácil de usar.
Características principales
- Interfaz segura con el sistema MiSeq FGx para el análisis automatizado de los datos posteriores a la secuenciación.
- Herramientas intuitivas para la gestión de muestras, la configuración de los ciclos, la visualización de datos y la elaboración de informes.
- Supervisión de los ensayos en tiempo real, comparaciones de muestras y gráficos de intensidad para una toma de decisiones informada.
- Preinstalado en un servidor dedicado para una computación potente sin complicaciones de infraestructura.
El software de análisis universal es una herramienta especializada que simplifica el análisis de datos genómicos forenses y ofrece una serie de funciones para analistas y laboratorios forenses.
TinyBio
Tinybio es una empresa pionera en IA genómica generativa que simplifica los procesos para los científicos reales con herramientas y software fáciles de usar. Se centra en mejorar la productividad y la optimización de recursos, permitiendo a los científicos concentrarse en su investigación sin complejidades de software.
Utiliza una herramienta de IA para comprender y sugerir posibles análisis sobre un conjunto de muestras. Tinybio es el más adecuado para quienes se dedican a la investigación genómica y buscan soluciones basadas en la IA para agilizar sus flujos de trabajo, desde la generación de experimentos hasta la construcción de pipelines de análisis.
Características principales
- Presenta un chatbot diseñado para idear, depurar y navegar por las complejidades de la bioinformática.
- Una herramienta especializada para construir rápidamente canalizaciones de análisis biológicos.
- Herramienta especializada para realizar ingeniería inversa del código responsable de la producción de un artículo científico.
- Proporciona una solución para organizar el entorno informático sin migración.
- Enfoque centrado en el usuario
El enfoque centrado en el usuario de la plataforma y su completo conjunto de herramientas la convierten en un valioso activo para bioinformáticos, científicos, biólogos computacionales e investigadores tanto del mundo académico como de la industria.
DeepVariant
DeepVariant, una tecnología de aprendizaje profundo, se desarrolló gracias a la colaboración entre el equipo de Google Brain y Verily Life Sciences. Esta innovadora herramienta está diseñada para afrontar el reto de reconstruir secuencias genómicas precisas y completas a partir de datos de secuenciación de alto rendimiento (HTS).
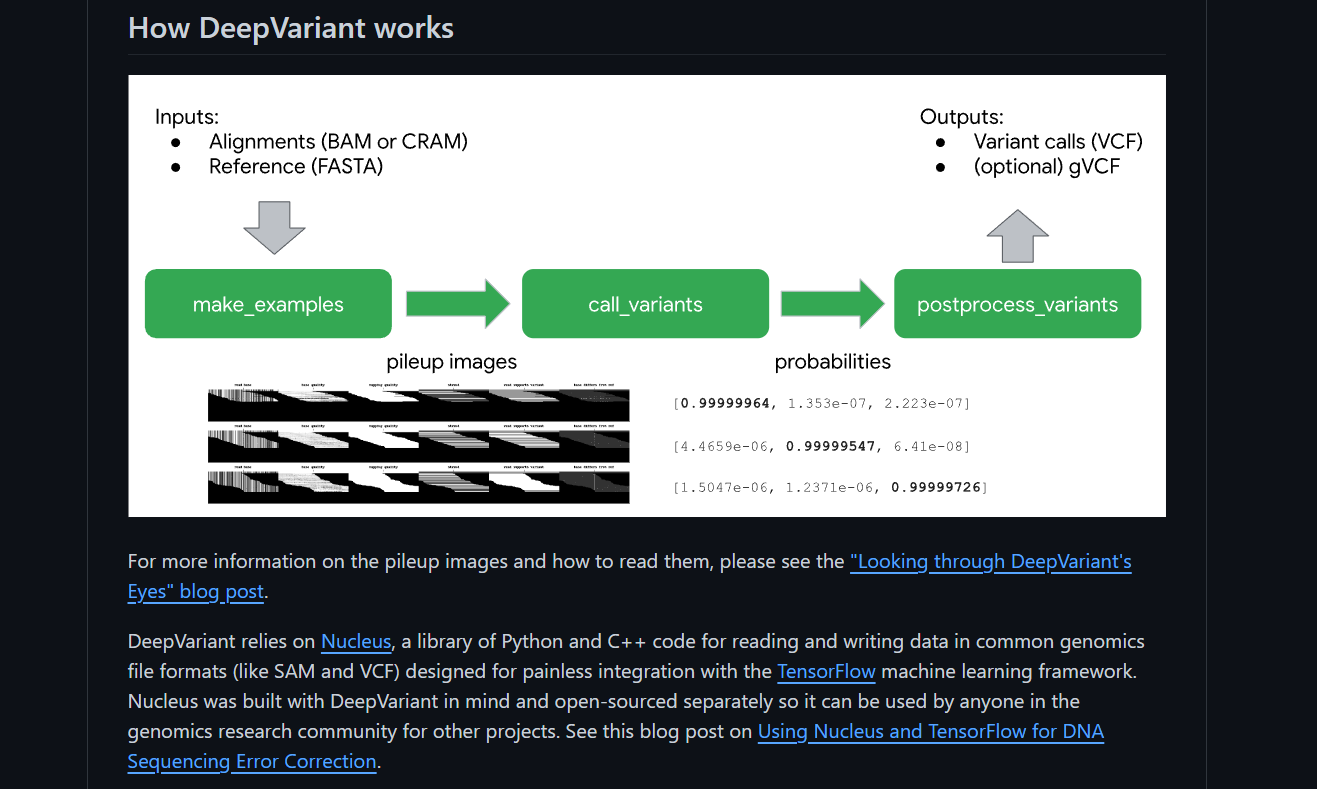
La HTS genera alrededor de 1.000 millones de secuencias cortas de ADN (reads), que representan sólo una fracción de todo el genoma. Se trata de una canalización de análisis de código abierto que utiliza una red neuronal profunda para llamar a las variantes genéticas a partir de datos de secuenciación de ADN de nueva generación.
Características principales
- DeepVariant aplica de forma única técnicas de aprendizaje profundo para abordar los retos de la llamada de variantes, ofreciendo una precisión mejorada en comparación con los métodos tradicionales.
- La transformación del problema de la llamada de variantes en un problema de clasificación de imágenes diferencia a DeepVariant.
- Asociación con GCP para desplegar los flujos de trabajo de DeepVariant en la nube
DeepVariant es una herramienta útil para la investigación genómica, que utiliza el aprendizaje profundo y es de código abierto para investigadores y bioinformáticos.
Galaxy
Galaxy es una plataforma web de código abierto para la investigación biomédica intensiva en datos. La utilizan los científicos para analizar grandes conjuntos de datos biomédicos como los de genómica, proteómica, metabolómica e imagen.
Con una interfaz gráfica, admite varios formatos de datos biológicos y es de código abierto. Sus aplicaciones incluyen la expresión génica, la proteómica y la transcriptómica, entre otras.
Características principales
- Ofrece miles de herramientas entre las que elegir
- Interfaz fácil de usar e interfaz gráfica intuitiva
- Galaxy admite una gran variedad de formatos de datos biológicos
- Proporciona CPU y espacio en disco suficientes para analizar grandes conjuntos de datos
La galaxia será útil para bioinformáticos, investigadores de fármacos y biólogos computacionales que pueden utilizarla desde que se aplica por la borda al campo de la bioinformática.
DNAnexus
DNAnexus, en genómica mediante el desarrollo de una plataforma basada en API dedicada a facilitar el intercambio y la gestión de datos y herramientas esenciales para acelerar la investigación genómica.
DNAnexus proporciona una red global para la genómica, que permite a científicos y médicos de todo el mundo colaborar de forma segura en investigaciones genómicas relacionadas con diversos campos, como el cáncer, las enfermedades cardiacas, la enfermedad de Alzheimer, las pruebas prenatales no invasivas y la agricultura.
Características principales
- Los productos de DNAnexus incluyen soluciones de gestión de datos, soluciones de análisis de datos, soluciones de colaboración, análisis de datos biomédicos y aplicaciones bioinformáticas de software.
- La plataforma funciona sobre una infraestructura basada en API, lo que simplifica la gestión de datos y herramientas y acelera los procesos de investigación genómica.
- DNAnexus aborda los retos de la informática genómica y la portabilidad de datos proporcionando una solución basada en la nube.
DNAnexus proporciona una plataforma segura, escalable y compatible para bioinformáticos, empresas e investigadores de todo el mundo.
Autodock
AutoDock es un conjunto de herramientas automatizadas de acoplamiento diseñadas para predecir cómo pequeñas moléculas, como candidatos a fármacos, se unen a un receptor con una estructura tridimensional conocida.
AutoDock es idóneo para aplicaciones en cristalografía de rayos X, diseño de fármacos basado en estructuras, optimización de pistas, cribado virtual, diseño de bibliotecas combinatorias, acoplamiento proteína-proteína y estudios de mecanismos químicos.

Características principales
- AutoDock facilita el acoplamiento de ligandos (moléculas pequeñas) a un conjunto de rejillas que representan la proteína diana.
- AutoDock 4 incluye auto dock para el acoplamiento de ligandos y autogrid para el cálculo previo de rejillas que describen la proteína diana.
- AutoDockTools (ADT) es una interfaz gráfica de usuario que facilita la configuración de enlaces rotatorios en el ligando y el análisis del acoplamiento.
- AutoDock Vina calcula internamente las cuadrículas de los tipos de átomos necesarios para el acoplamiento, lo que elimina la necesidad de que los usuarios elijan los tipos de átomos y calculen previamente los mapas de cuadrículas.
La versatilidad de AutoDock lo convierte en una valiosa herramienta para una amplia gama de aplicaciones científicas, especialmente en los sectores de las ciencias de la vida y el desarrollo de fármacos. Es de gran ayuda para investigadores y biólogos.
Rosetta
El paquete de software Rosetta engloba algoritmos para el modelado computacional y el análisis de estructuras de proteínas. Su aplicación ha dado lugar a importantes avances en biología computacional, facilitando logros como el diseño de novo de proteínas, el diseño de enzimas, el acoplamiento de ligandos y la predicción de estructuras de macromoléculas y complejos biológicos.

Características principales
- Comprensión de las interacciones macromoleculares
- Diseño de moléculas a medida
- Creación de métodos eficaces para explorar el espacio de conformaciones y secuencias
- Encontrar funciones de energía ampliamente útiles para diversas representaciones biomoleculares
El conjunto de herramientas de Rosetta es útil para científicos, biólogos computacionales, estudiantes e investigadores dedicados al modelado macromolecular y la biología estructural.
BioJava
BioJava es una plataforma bioinformática especializada diseñada para el entorno Java, que satisface las necesidades de procesamiento de diversos datos biológicos.
La plataforma ejecuta diversas operaciones, como la manipulación de secuencias, el análisis de estructuras de proteínas, la utilización del Sistema de Anotación Distribuida (DAS) y la programación dinámica, y admite la interoperabilidad con la arquitectura CORBA (Common Object Request Broker Architecture).
Características principales
- Las capacidades de BioJava incluyen la gestión de instalaciones PDB locales, la manipulación de estructuras, la realización de análisis estándar como la alineación de secuencias y estructuras, y la visualización en 3D.
- Facilita la recuperación de datos de bases de datos de secuencias de nucleótidos y proteínas.
- BioJava también admite tareas como la lectura y escritura de formatos de archivos de secuencias, la traducción de secuencias de ADN a proteínas y la ejecución de rutinas bioinformáticas comunes.
- Permite la búsqueda de secuencias similares y la manipulación de secuencias individuales.
BioJava es muy adecuado para personas e investigadores del campo de la bioinformática, científicos y biólogos computacionales que prefieren utilizar herramientas basadas en Java para procesar diversos datos biológicos.
Bionano
Las soluciones de Bionano ofrecen una resolución sin precedentes en todas las clases de variación genómica, abordando las limitaciones de las herramientas y métodos tradicionales. El software de análisis de Bionano (aplicaciones de inteligencia de variantes) integra los datos OGM con NGS, microarrays cromosómicos (CMA) y otros tipos de datos, lo que permite una interpretación y un análisis exhaustivos de los datos de variantes. Las áreas de interés de Bionano abarcan la atención clínica, la investigación y la terapéutica.

Características principales
- El software de análisis VIA utiliza los datos de microarrays y NGS para evaluar el DRH
- Simplifica el ensamblaje del genoma, el análisis de variantes estructurales y el andamiaje híbrido
- El sistema Saphyr detecta y analiza las variantes estructurales con gran velocidad y rendimiento
- Nexus Copy Number permite el análisis visual y estadístico de la variación genética en cohortes de investigación.
Es una herramienta inestimable para investigadores genómicos, clínicos y biotecnólogos que pretendan ampliar los límites de la comprensión y la aplicación de la genómica.
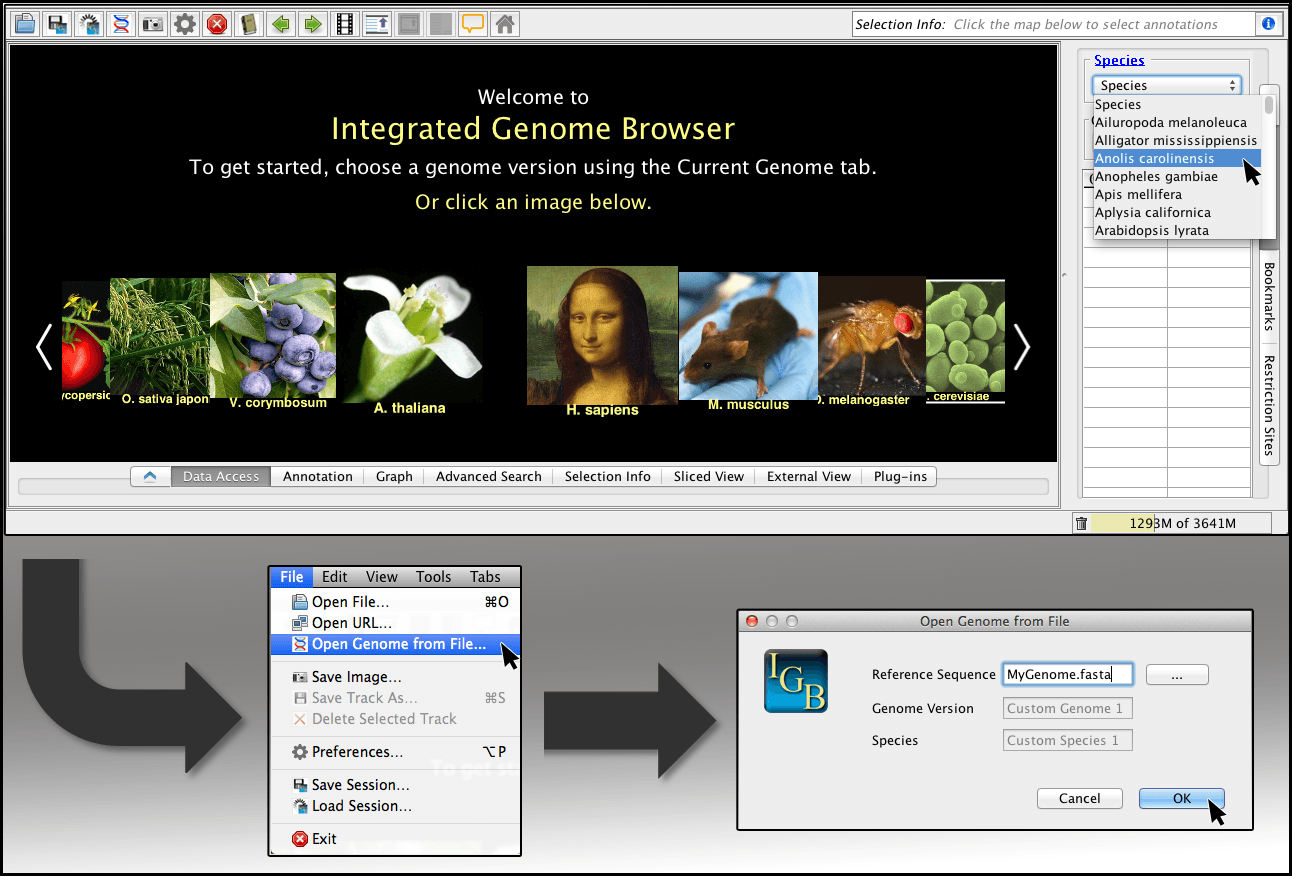
El Navegador genómico integrado sirve como herramienta de visualización diseñada para ilustrar intrincados patrones biológicos dentro de conjuntos de datos genómicos, datos de secuencias, modelos de genes y datos de micromatrices de ADN.
Compatible con los sistemas operativos UNIX, Linux, Mac y Windows, este software ofrece una solución fiable y rápida para visualizar grandes cantidades de datos directamente en un escritorio.
Características principales
- Agilice su flujo de trabajo empleando scripts para definir un genoma. También puede utilizar R para gestionar IGB.
- Elabore, personalice y guarde gráficos a partir de sus datos. Utilice gráficos de profundidad para mostrar la cobertura o gráficos de desajuste para contar las diferencias entre sus datos y una referencia.
- También puede hacer sus plug-ins/apps y compartirlos con la comunidad IGB.
- Puede crear un IGB QuickLoad para compartir datos entre colaboradores. Almacene sus datos en la nube para facilitar el acceso a todos los implicados en el proyecto.
El Navegador Integrado del Genoma es una herramienta útil para expertos en datos, científicos y bioinformáticos para automatizar el flujo de trabajo.
DRAGEN
El análisis DRAGEN encuentra aplicaciones en diversos campos de las ciencias biológicas, ofreciendo un enfoque transformador del análisis genómico. El análisis DRAGEN proporciona un examen secundario preciso, exhaustivo y racionalizado de los datos de secuenciación de nueva generación.
El análisis DRAGEN, que aprovecha la tecnología FPGA altamente reconfigurable, acelera los algoritmos de análisis genómico al tiempo que aborda los retos que plantean los tiempos de cálculo y los volúmenes de datos.
Características principales
- Analiza genomas completos, exomas, metilomas y transcriptomas utilizando una plataforma unificada.
- Utiliza el genoma gráfico de referencia y el aprendizaje automático, impulsando una precisión sin precedentes.
- Identifique y perfile enfermedades infecciosas con una solución integral.
- El análisis DRAGEN proporciona la flexibilidad necesaria para insertar una variedad de archivos de entrada y producir una gama de documentos de salida.
El análisis DRAGEN resulta muy beneficioso para el análisis genómico. Es útil para biólogos computacionales y analistas genómicos.
PathAI
PathAI aprovecha la visión por ordenador y el aprendizaje profundo para analizar imágenes de patología, transformando el enfoque tradicional del diagnóstico y el tratamiento del cáncer.
Las tres aplicaciones principales de los productos de PathAI incluyen ayudar a los patólogos a realizar mejores diagnósticos, mejorar el desarrollo de fármacos mediante un uso terapéutico informado y ampliar los servicios de patología de referencia a regiones que carecen de acceso.

Características principales
- Automatiza las tareas monótonas de los patólogos, mejorando la eficiencia en el análisis de los portaobjetos de patología.
- Integra a la perfección la histopatología con los datos genómicos, proporcionando información valiosa para fundamentar las decisiones terapéuticas.
- Utiliza una variedad de datos y resultados, junto con múltiples escáneres, tinciones y fuentes de laboratorio, para hacer mejores predicciones evitando sesgos.
Al combinar la experiencia de los patólogos con el software de PathAI, la plataforma libera tiempo y permite centrarse en determinar las opciones de tratamiento óptimas para cada paciente.
Geneious
El software Geneious, desarrollado en Java Swing, cuenta con un alto nivel de interoperabilidad en los sistemas operativos más utilizados. Sirve como herramienta integral de análisis biológico con una interfaz fácil de usar.
Soporta múltiples funcionalidades como alineamientos múltiples, árboles filogenéticos, ensamblajes contig, gráficos estadísticos, estructuras 3D, cromatogramas y electroferogramas.

Características principales
- Ensamblaje y análisis de secuenciación de próxima generación
- Visualizaciones y gráficos
- Geneious permite la recuperación de bases de datos biológicas de varias plataformas.
- Edición y ensamblaje de cromatogramas
- Alineación y Filogenética
Geneious es un software bioinformático ampliamente conocido, útil para investigadores, bioinformáticos y biólogos computacionales.
MEGA
MEGA, abreviatura de Molecular Evolutionary Genetics Analysis, es un completo paquete de software creado para la exploración y visualización de datos de secuencias moleculares. Su objetivo principal es la investigación de las relaciones evolutivas incrustadas en las secuencias de ADN o proteínas.
MEGA dota a los investigadores de herramientas versátiles que les permiten realizar análisis filogenéticos, estimar distancias evolutivas y construir árboles filogenéticos detallados.
Características principales
- MEGA permite a los usuarios construir árboles filogenéticos para visualizar y analizar las relaciones evolutivas entre secuencias biológicas.
- Varios enfoques estadísticos están integrados en MEGA para evaluar la importancia de los resultados en los análisis de evolución molecular.
- MEGA proporciona herramientas de visualización para explorar e interpretar datos moleculares complejos, incluidos los alineamientos de secuencias y los árboles filogenéticos.
Investigadores, bioinformáticos y científicos utilizan MEGA para obtener información sobre la historia evolutiva de genes, proteínas y otras entidades biológicas.
BLAST
La herramienta de búsqueda de alineaciones locales básicas (BLAST) identifica similitudes locales entre secuencias comparando secuencias de nucleótidos o proteínas con bases de datos. Calcula la significación estadística de las coincidencias, lo que permite inferir relaciones funcionales y evolutivas entre secuencias y ayuda a identificar a los miembros de una familia de genes.
BLAST es un programa y un algoritmo bioinformático muy utilizado para comparar información de secuencias biológicas primarias, como las secuencias de aminoácidos de las proteínas o los nucleótidos de las secuencias de ADN.
Características principales
- BLAST se utiliza principalmente para comparar una secuencia de consulta con una base de datos de secuencias para encontrar secuencias similares u homólogas.
- Existen varias versiones de BLAST, entre las que se incluyen BLAST de nucleótidos (para secuencias de nucleótidos), BLAST de proteínas (para secuencias de proteínas), BLASTx (traduce las secuencias de nucleótidos a secuencias de proteínas antes de compararlas) y otras.
- BLAST puede realizar búsquedas en varias bases de datos biológicas, incluidas las bases de datos de nucleótidos y proteínas del NCBI.
BLAST es una herramienta vital en bioinformática y biología molecular, esencial para identificar con rapidez y precisión similitudes en secuencias de ADN, ARN y proteínas.
Clara Parabricks
Clara Parabricks es una suite de software rentable para el análisis secundario rápido de datos de ADN y ARN de NGS, que ofrece resultados excepcionalmente rápidos en comparación con otros métodos. Como herramienta integral de análisis genómico, Parabricks mejora significativamente el tiempo de procesamiento para tareas como el análisis de la línea germinal y somática.
Por ejemplo, puede analizar 30x de datos del genoma humano completo en sólo 25 minutos, una mejora notable respecto a las 30 horas que se requieren habitualmente. Su salida se alinea con el software ampliamente utilizado, lo que garantiza una fácil verificación de los resultados.
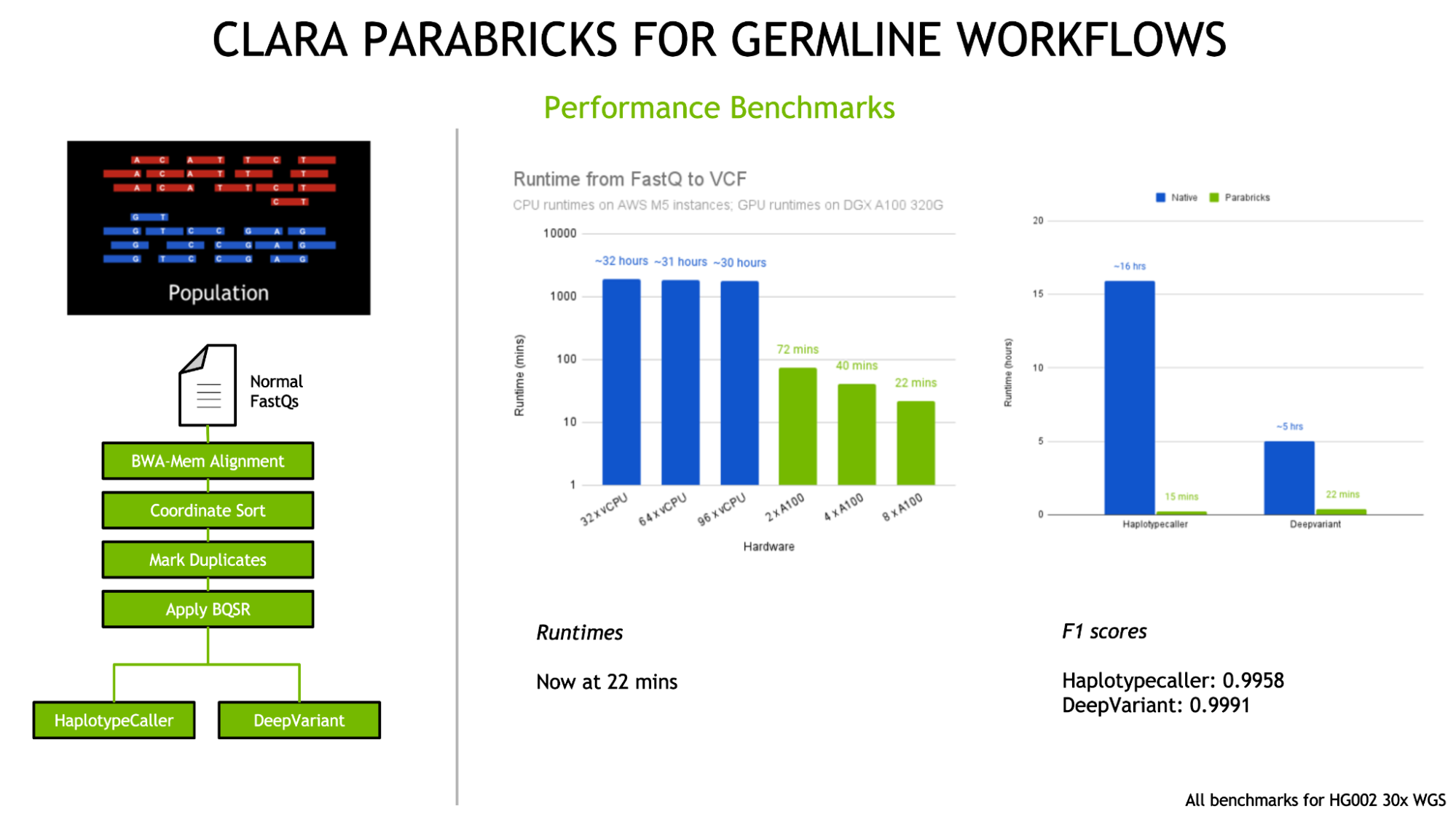
Características clave
- Clara Parabricks se acelera exclusivamente en la GPU, lo que optimiza el análisis secundario con notable rapidez
- Integración de herramientas basadas en el aprendizaje profundo junto con las estándar del sector.
- Reducción significativa del tiempo de cálculo y de los costes de análisis.
- Compatibilidad con Azure y disponibilidad gratuita en NGC.
- Versatilidad para soportar diversos flujos de trabajo genómicos.
Clara Parabricks es ideal para investigadores genómicos, bioinformáticos, laboratorios clínicos, empresas farmacéuticas, instituciones educativas y profesionales sanitarios.
Pluto
Pluto es una plataforma versátil y fácil de usar que está revolucionando la investigación científica. Simplifica la gestión de proyectos, permitiendo a los equipos organizar, supervisar y asignar experimentos con un intercambio seguro de datos para una colaboración eficaz.
Los científicos se benefician de sus sólidas herramientas de análisis y visualización de datos, que admiten algoritmos de biología computacional para obtener información sobre la expresión génica, la unión ADN-proteína y los biomarcadores.
Características principales
- Capacitación de los científicos para ejecutar potentes análisis bioinformáticos directamente en el navegador.
- Transformación de datos brutos en gráficos listos para su publicación en cuestión de minutos.
- Plataforma elegante para organizar, supervisar y asignar experimentos.
- Herramientas intuitivas para analizar y visualizar datos biológicos complejos.
- Compatibilidad con diversos ensayos biológicos, incluidos RNA-seq, ChIP-seq, CUT&RUN y ensayos basados en placas.
Pluto es una plataforma versátil diseñada para usuarios en investigación biológica, biología computacional, gestión de proyectos y esfuerzos científicos colaborativos. Con su interfaz fácil de usar y sus completas funciones, tiene valor para diversas funciones dentro de la comunidad de investigación científica.
LatchBio
Latchbio es la plataforma más flexible de acceso y análisis de datos para la I+D biológica. Es una nube biológica donde las organizaciones pueden almacenar, procesar, analizar y visualizar datos multiómicos.
Puede cargar flujos de trabajo bioinformáticos en cualquier idioma utilizando el SDK de Latch, recibir interfaces asociadas sin código para sus científicos y beneficiarse de una infraestructura altamente escalable que da soporte a toda su empresa.

Características principales
- Latch Registry es una base de datos fácil de usar con una interfaz de hoja de cálculo diseñada para facilitar la captura de metadatos intrincados para archivos de secuenciación de próxima generación (NGS) y multiómica dentro de la plataforma Latch.
- Latch Data es un sistema versátil de almacenamiento de archivos capaz de alojar datos ilimitados y proporcionar acceso universal a todos los miembros de su organización a través de un inicio de sesión unificado.
- Los Latch Pods destacan como unidades de computación en la nube ágiles y robustas, y cuentan con RStudio y JupyterLab preinstalados para un análisis posterior sin esfuerzo de los resultados del flujo de trabajo.
Latchbio le permite acceder a sus datos y analizarlos con una interfaz fácil de usar. Por lo tanto, es una herramienta útil para bioinformáticos, científicos de laboratorio húmedo y jefes de equipo.
Conclusión
Las herramientas bioinformáticas son esenciales para comprender y desentrañar las complejidades de los datos biológicos. Desde la organización de la información hasta la predicción de las estructuras de las proteínas y la realización de análisis genómicos, estas herramientas benefician por igual a investigadores, científicos y estudiantes.
A medida que la tecnología mejora, el trabajo en equipo entre la biología y la informática sigue ayudándonos a aprender más sobre la vida. Las herramientas mencionadas en este artículo le ayudarán a analizar datos biológicos, realizar análisis estadísticos, visualizar datos, llamar variantes, predecir estructuras de proteínas, utilizar navegadores genómicos, realizar alineaciones de secuencias y mucho más.