La bioinformatique est un domaine scientifique qui associe la biologie et l’informatique pour analyser les données biologiques. Les outils bioinformatiques permettent d’organiser, de comparer et d’analyser les données, de prédire les structures des protéines, d’effectuer des analyses génomiques et de soutenir la recherche.
Dans cet article, j’ai rassemblé quelques outils qui seront utiles non seulement aux bioinformaticiens, mais aussi aux chercheurs, aux scientifiques, aux analystes, aux étudiants, etc.
Je parlerai de divers outils qui peuvent vous aider à analyser des données biologiques, à effectuer des analyses statistiques, à visualiser des données, à appeler des variants, à prédire des structures de protéines, à utiliser des navigateurs de génome, à effectuer des alignements de séquences, et bien plus encore.

OmicsBox
OmicsBox est un logiciel de bioinformatique de premier ordre, qui permet une analyse approfondie des données de séquençage de nouvelle génération (NGS) pour les génomes, les transcriptomes et les métagénomes. Il est largement utilisé par des institutions de recherche de premier plan dans le monde entier et a été conçu pour être convivial, efficace et doté d’outils puissants pour traiter des ensembles de données complexes et volumineux.
OmicsBox est structuré en modules, chacun adapté à des analyses spécifiques, y compris l’analyse du génome, la variation génétique, la transcriptomique, l’analyse fonctionnelle et la métagénomique.

Caractéristiques principales
- Le module d’analyse du génome est un ensemble d’outils efficaces et conviviaux permettant de caractériser et d’analyser les génomes nouvellement séquencés.
- Le module de transcriptomique permet de traiter les données RNA-Seq depuis les lectures brutes jusqu’à l’analyse fonctionnelle de manière flexible et intuitive.
- Le module d’analyse fonctionnelle fournit un contexte biologique en tant qu’option d’analyse.
- Le module métagénomique permet l’analyse des données microbiomiques, y compris l’assemblage, l’annotation et la classification des données métagénomiques.
OmicsBox fournit une suite d’outils pour l’analyse des données génomiques, transcriptomiques et métagénomiques, ce qui en fait un outil précieux pour les professionnels de la biologie computationnelle et de la bioinformatique.
Bioconductor
Bioconductor est un logiciel libre qui comprend des paquets R spécialisés dans l’interprétation des données génomiques à haut débit. Le langage et l’environnement R pour le calcul statistique et les graphiques sont améliorés par Bioconductor pour répondre aux défis uniques posés par la génomique et la bioinformatique.
La boîte à outils complète fournie par Bioconductor permet aux utilisateurs d’effectuer toute une série de tâches, du prétraitement de base des données aux analyses statistiques avancées.
Caractéristiques principales
- La boîte à outils Bioconductor fournit une vaste gamme de paquets R spécialisés pour des tâches telles que l’analyse de données de microréseaux, de données de séquençage à haut débit, de données de cytométrie de flux, et bien plus encore.
- Documentation de haute qualité et recherche reproductible
- Les paquets de Bioconductor comprennent souvent des fonctionnalités permettant de s’interfacer avec des bases de données génomiques populaires, telles qu’Ensembl et UCSC Genome Browser, et d’en extraire des données.
La boîte à outils Bioconductor est largement utilisée par les bioinformaticiens, les chercheurs et les scientifiques qui travaillent avec des données génomiques à haut débit.
FastQC
FastQC est un outil de contrôle de la qualité très répandu, conçu pour les données de séquence générées par les méthodes de séquençage de nouvelle génération. FastQC offre une approche simple des contrôles de qualité sur les données brutes des pipelines de séquençage.
L’outil est facilement téléchargeable et présente une interface conviviale, permettant aux utilisateurs de résoudre les problèmes liés aux données avant de poursuivre l’analyse. Les fichiers d’entrée consistent en des séquences de lecture, et l’outil génère des sorties sous forme de graphiques et de résumés tabulaires des résultats.
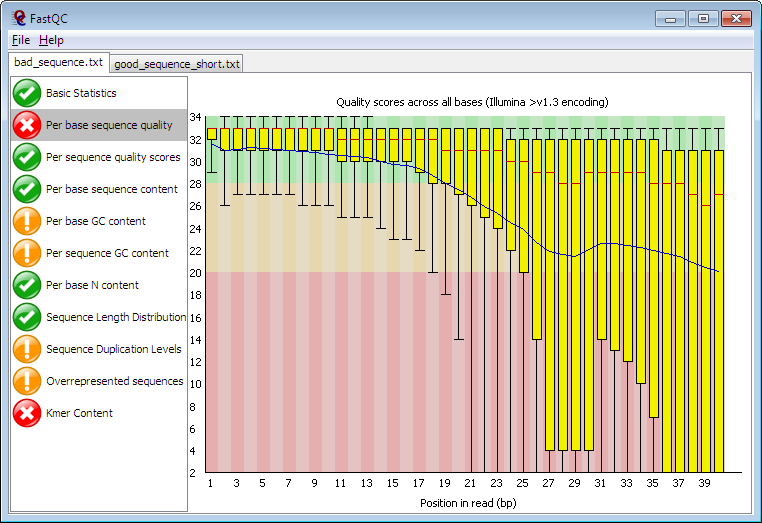
Caractéristiques de Kay
- Les rapports peuvent être générés automatiquement sans utiliser l’application interactive lorsqu’ils sont hors ligne.
- Ils fournissent une vue d’ensemble rapide du problème.
- Importation de données à partir de fichiers BAM, SAM ou FastQ (toute variante) pour une analyse pratique.
- Génère des graphiques et des tableaux récapitulatifs pour l’évaluation de la qualité des données
- Exportation de rapports HTML
Ce logiciel est idéal pour le traitement et l’analyse des données de séquençage. Il est donc recommandé aux analystes de données biologiques, aux chercheurs et aux équipes de projet.
EMBOSS
EMBOSS (European Molecular Biology Open Software Suite) est l’un des outils pour apprendre l’analyse bioinformatique si vous êtes débutant. EMBOSS est un logiciel de pointe, gratuit et libre, méticuleusement conçu pour répondre aux exigences uniques de la communauté de la biologie moléculaire et de la bioinformatique.
EMBOSS dispose de nombreux outils pour analyser les séquences, et il fonctionne bien avec d’autres progiciels et outils populaires. L’un d’entre eux est EMBOSS Needle :
Caractéristiques principales
EMBOSS comprend plus de 200 applications pour
- Montrer les caractéristiques d’une séquence
- Recherche dans les bases de données
- Présentation des données de séquences
- modèles moléculaires en 3D
- Plateformes bioinformatiques
- Décomposition d’une séquence en séquences plus petites
Il s’agit d’un outil convivial pour les étudiants, les chercheurs en informatique, les bioinformaticiens et les scientifiques des données.
Clustal
Clustal est un ensemble d’outils bioinformatiques tels que ClustalX, ClustalW et Clustal Omega. Il est conçu pour aligner des séquences biologiques telles que l’ADN, l’ARN et les protéines. Ils permettent d’identifier les régions similaires dans plusieurs séquences, ce qui facilite l’analyse des relations évolutives et des domaines fonctionnels.
La série de programmes Clustal est largement utilisée en biologie moléculaire pour l’alignement multiple de séquences d’acides nucléiques et de protéines et pour la préparation d’arbres phylogénétiques.
Caractéristiques principales
- Le logiciel Clustal offre généralement des interfaces conviviales
- Clustal excelle dans l’alignement de trois séquences ou plus
- Clustal X permet de prédire l’alignement de séquences multiples et l’analyse phylogénétique pour des séquences génétiques données de divers organismes.
- Clustal Omega utilise une heuristique basée sur l’analyse phylogénétique
Clustal est particulièrement adapté aux professionnels de la bioinformatique et de la biologie computationnelle qui doivent aligner et analyser de multiples séquences biologiques.
DNASTAR Lasergene
DNASTAR Lasergene est une suite logicielle de pointe qui constitue une solution polyvalente pour la biologie moléculaire, la génomique et l’analyse des protéines. Cette plateforme complète est utilisée par des scientifiques du monde entier.
De plus, DNASTAR offre les applications Nova, incluant NovaFold AI, NovaFold, NovaFold Antibody, et NovaDock, améliorant ainsi ses capacités de prédiction et d’analyse de la structure des protéines.

Caractéristiques principales
- Solutions complètes pour la biologie moléculaire, les anticorps, la génomique et l’analyse des protéines, offrant une suite complète pour les chercheurs.
- Automatise les tâches dans les projets de génomique, en rationalisant l’assemblage et l’analyse des séquences.
- Fournit des représentations graphiques 3D flexibles et riches des structures protéiques, améliorant la compréhension des séquences protéiques.
DNASTAR Lasergene, avec ses packages complets et ses applications Nova avancées, est une suite logicielle polyvalente qui répond aux divers besoins des biologistes moléculaires, des chercheurs en génomique, des protéinologues et des bioinformaticiens structurels.
GATK
Le Genome Analysis Toolkit (GATK), créé par la plateforme Data Science du Broad Institute, est un outil puissant pour la découverte de variantes génétiques et le génotypage. Il excelle dans le traitement de fichiers d’entrée volumineux et se concentre sur l’identification de variations telles que les SNP et les indels dans les données d’ADN et d’ARN-Seq.
GATK traite également les nombres de copies et les variations structurelles et inclut des utilitaires pour le contrôle de la qualité dans le séquençage à haut débit.

Caractéristiques principales
- GATK est optimisé pour obtenir des résultats précis et de haute qualité.
- GATK utilise un cadre de programmation structuré, tirant parti de la philosophie de programmation fonctionnelle de MapReduce.
- Il maximise l’efficacité des calculs pour un traitement efficace.
- Il est capable de réaliser des analyses génomiques pour les exomes et les génomes entiers.
- Il propose des flux de travail fondés sur les meilleures pratiques pour les variants somatiques courts.
GATK est un outil destiné à ceux qui souhaitent naviguer dans les complexités de l’analyse génomique avec précision et facilité, tels que les chercheurs en génomique, les scientifiques et les bioinformaticiens.
Logiciel d’analyse universel
Le logiciel d’analyse universel (UAS) simplifie l’analyse et la gestion des données génomiques médico-légales. Il prend en charge divers flux de travail ForenSeq, traite rapidement les données et fournit des appels de variants fiables sans nécessiter de licences par siège.
Conçu pour les analystes médico-légaux, le logiciel UAS rationalise le traitement des informations sur les séquences, facilitant ainsi l’examen efficace des profils ADN. Le système UAS est conçu pour être compatible avec le système de séquençage MiSeq FGx et les outils tiers courants, ce qui en fait une solution conviviale et polyvalente.
Caractéristiques principales
- Interface sécurisée avec le système MiSeq FGx pour l’analyse automatisée des données de post-séquençage.
- Outils intuitifs pour la gestion des échantillons, la configuration de l’exécution, la visualisation des données et la production de rapports.
- Surveillance de l’exécution en temps réel, comparaisons d’échantillons et tracés d’intensité pour une prise de décision éclairée.
- Préinstallé sur un serveur dédié pour une puissance de calcul sans souci d’infrastructure.
Universal Analysis Software est un outil spécialisé qui simplifie l’analyse des données génomiques médico-légales, offrant une gamme de fonctionnalités pour les analystes médico-légaux et les laboratoires.
TinyBio
Tinybio est une entreprise pionnière dans le domaine de l’IA générative génomique qui simplifie les processus pour les scientifiques réels grâce à des outils et des logiciels conviviaux. Elle se concentre sur l’amélioration de la productivité et l’optimisation des ressources, permettant aux scientifiques de se concentrer sur leurs recherches sans avoir à se soucier de la complexité des logiciels.
Tinybio utilise un outil d’IA pour comprendre et suggérer des analyses possibles sur un ensemble d’échantillons. Tinybio convient parfaitement aux chercheurs en génomique qui recherchent des solutions basées sur l’IA pour rationaliser leurs flux de travail, de la génération d’expériences à la construction de pipelines d’analyse.
Caractéristiques principales
- Comprend un chatbot conçu pour concevoir, déboguer et naviguer dans les complexités de la bioinformatique.
- Un outil spécialisé pour construire rapidement des pipelines d’analyse biologique.
- Un outil spécialisé pour l’ingénierie inverse du code responsable de la production d’un article scientifique.
- Fournit une solution pour organiser l’environnement informatique sans migration.
- Approche centrée sur l’utilisateur
L’approche centrée sur l’utilisateur et la suite d’outils complète de la plateforme en font un atout précieux pour les bioinformaticiens, les scientifiques, les biologistes computationnels et les chercheurs, tant dans les universités que dans l’industrie.
DeepVariant
DeepVariant, une technologie d’apprentissage en profondeur, a été développée grâce à la collaboration de l’équipe Google Brain et de Verily Life Sciences. Cet outil innovant est conçu pour relever le défi de la reconstruction de séquences génomiques précises et complètes à partir de données de séquençage à haut débit (HTS).
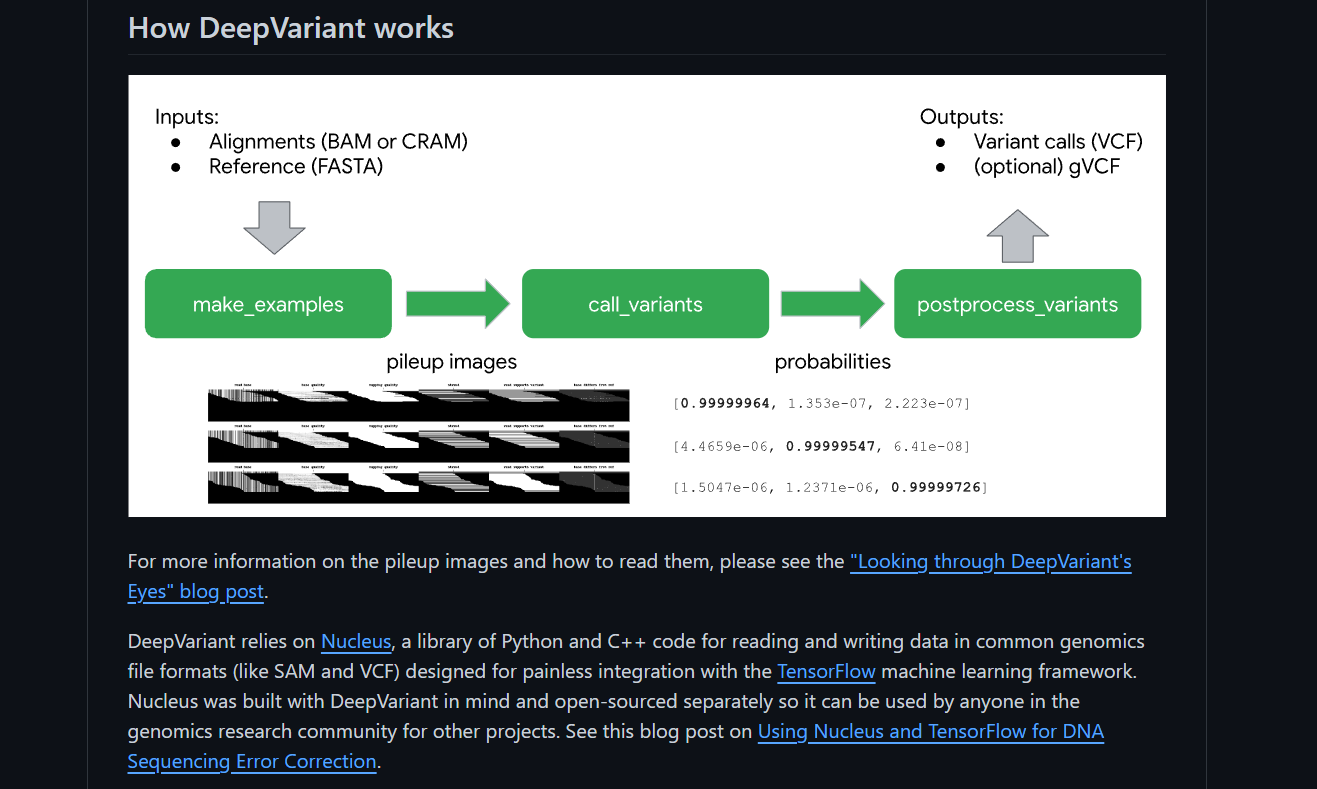
Le séquençage à haut débit génère environ 1 milliard de courtes séquences d’ADN (reads), qui ne représentent qu’une fraction du génome entier. Il s’agit d’un pipeline d’analyse open-source qui utilise un réseau neuronal profond pour appeler des variantes génétiques à partir de données de séquençage d’ADN de nouvelle génération.
Caractéristiques principales
- DeepVariant applique de manière unique des techniques d’apprentissage profond pour relever les défis de l’appel de variants, offrant une précision améliorée par rapport aux méthodes traditionnelles.
- DeepVariant se distingue par la transformation du problème de l’appel de variants en un problème de classification d’images.
- Partenariat avec GCP pour déployer les flux de travail de DeepVariant sur le cloud
DeepVariant est un outil utile pour la recherche génomique, utilisant l’apprentissage profond et étant open-source pour les chercheurs et les bioinformaticiens.
Galaxy
Galaxy est une plateforme web à code source ouvert pour la recherche biomédicale à forte intensité de données. Elle est utilisée par les scientifiques pour analyser de grands ensembles de données biomédicales comme celles de la génomique, de la protéomique, de la métabolomique et de l’imagerie.
Doté d’une interface graphique, il prend en charge divers formats de données biologiques et est open-source. Ses applications comprennent l’expression des gènes, la protéomique, la transcriptomique, etc.
Caractéristiques principales
- Il propose des milliers d’outils au choix
- Interface conviviale et interface graphique intuitive
- Galaxy prend en charge une grande variété de formats de données biologiques
- L’unité centrale et l’espace disque sont suffisants pour analyser de grands ensembles de données
Galaxy sera utile aux bioinformaticiens, aux chercheurs de médicaments et aux biologistes computationnels qui peuvent l’utiliser depuis qu’il a été appliqué au domaine de la bioinformatique.
DNAnexus
DNAnexus, dans le domaine de la génomique, développe une plateforme basée sur une API destinée à faciliter le partage et la gestion des données et des outils essentiels à l’accélération de la recherche génomique.
DNAnexus fournit un réseau mondial pour la génomique, permettant aux scientifiques et aux cliniciens du monde entier de collaborer en toute sécurité à la recherche génomique dans divers domaines, notamment le cancer, les maladies cardiaques, la maladie d’Alzheimer, les tests prénataux non invasifs et l’agriculture.
Caractéristiques principales
- Les produits de DNAnexus comprennent des solutions de gestion des données, des solutions d’analyse des données, des solutions de collaboration, des analyses de données biomédicales et des applications logicielles de bio-informatique.
- La plateforme fonctionne sur une infrastructure basée sur l’API, ce qui simplifie la gestion des données et des outils et accélère les processus de recherche génomique.
- DNAnexus relève les défis de l’informatique génomique et de la portabilité des données en proposant une solution basée sur l’informatique en nuage.
DNAnexus fournit une plateforme sécurisée, évolutive et conforme aux bioinformaticiens, aux entreprises et aux chercheurs du monde entier.
Autodock
AutoDock est une suite d’outils d’amarrage automatisés conçus pour prédire comment les petites molécules, telles que les médicaments candidats, se lient à un récepteur dont la structure 3D est connue.
AutoDock est idéal pour les applications de cristallographie aux rayons X, la conception de médicaments basée sur la structure, l’optimisation des têtes de série, le criblage virtuel, la conception de bibliothèques combinatoires, l’amarrage protéine-protéine et les études de mécanismes chimiques.

Caractéristiques principales
- AutoDock facilite l’arrimage de ligands (petites molécules) à un ensemble de grilles représentant la protéine cible.
- AutoDock 4 comprend auto dock pour l’ancrage des ligands et autogrid pour le pré-calcul des grilles décrivant la protéine cible.
- AutoDockTools (ADT) est une interface utilisateur graphique qui facilite la configuration des liaisons rotatives dans le ligand et l’analyse de l’amarrage.
- AutoDock Vina calcule en interne les grilles pour les types d’atomes nécessaires à l’amarrage, ce qui évite aux utilisateurs de choisir les types d’atomes et de précalculer les cartes de grilles.
La polyvalence d’AutoDock en fait un outil précieux pour un large éventail d’applications scientifiques, en particulier dans les secteurs des sciences de la vie et du développement de médicaments. Il est utile aux chercheurs et aux biologistes.
Rosetta
La suite logicielle Rosetta comprend des algorithmes pour la modélisation informatique et l’analyse des structures protéiques. Son application a conduit à des percées significatives en biologie informatique, facilitant des réalisations telles que la conception de protéines de novo, la conception d’enzymes, l’amarrage de ligands et la prédiction de la structure de macromolécules et de complexes biologiques.

Caractéristiques principales
- Compréhension des interactions macromoléculaires
- Concevoir des molécules personnalisées
- Création de méthodes efficaces pour explorer l’espace des conformations et des séquences.
- Trouver des fonctions énergétiques largement utiles pour diverses représentations biomoléculaires
La boîte à outils de Rosetta est utile aux scientifiques, aux biologistes computationnels, aux étudiants et aux chercheurs engagés dans la modélisation macromoléculaire et la biologie structurelle.
BioJava
BioJava est une plateforme bioinformatique spécialisée, conçue pour l’environnement Java, qui répond aux besoins de traitement de diverses données biologiques.
La plateforme exécute diverses opérations telles que la manipulation de séquences, l’analyse de la structure des protéines, l’utilisation du système d’annotation distribué (DAS) et la programmation dynamique. Elle prend en charge l’interopérabilité avec l’architecture CORBA (Common Object Request Broker Architecture).

Caractéristiques principales
- Les capacités de BioJava comprennent la gestion des installations PDB locales, la manipulation des structures, la réalisation d’analyses standard telles que l’alignement des séquences et des structures, et la visualisation en 3D.
- Il facilite la recherche de données dans les bases de données pour les séquences de nucléotides et de protéines.
- BioJava prend également en charge des tâches telles que la lecture et l’écriture de formats de fichiers de séquences, la traduction de séquences d’ADN en protéines et l’exécution de routines bioinformatiques courantes.
- Il permet de rechercher des séquences similaires et de manipuler des séquences individuelles.
BioJava est bien adapté aux personnes et aux chercheurs dans le domaine de la bio-informatique, aux scientifiques et aux biologistes informatiques qui préfèrent utiliser des outils basés sur Java pour traiter diverses données biologiques.
Bionano
Les solutions de Bionano offrent une résolution inégalée pour toutes les classes de variations génomiques, en s’attaquant aux limites des outils et méthodes traditionnels. Le logiciel d’analyse de Bionano (Variant Intelligence Applications) intègre les données OGM avec les données NGS, les puces chromosomiques (CMA) et d’autres types de données, ce qui permet une interprétation et une analyse complètes des données de variants. Les domaines d’intérêt de Bionano couvrent les soins cliniques, la recherche et la thérapeutique.

Caractéristiques principales
- Le logiciel d’analyse VIA utilise des données de microréseaux et de NGS pour évaluer la RHD
- Simplifie l’assemblage du génome, l’analyse des variantes structurelles et l’échafaudage hybride
- Le système Saphyr détecte et analyse les variants structurels à grande vitesse et à haut débit
- Nexus Copy Number permet l’analyse visuelle et statistique des variations génétiques dans les cohortes de recherche.
Il s’agit d’un outil précieux pour les chercheurs en génomique, les cliniciens et les biotechnologues qui cherchent à repousser les limites de la compréhension et de l’application de la génomique.
L’Integrated Genome Browser est un outil de visualisation conçu pour illustrer les schémas biologiques complexes dans les ensembles de données génomiques, les données de séquences, les modèles de gènes et les données de puces à ADN.
Compatible avec les systèmes d’exploitation UNIX, Linux, Mac et Windows, ce logiciel offre une solution fiable et rapide pour la visualisation de données volumineuses directement sur un bureau.
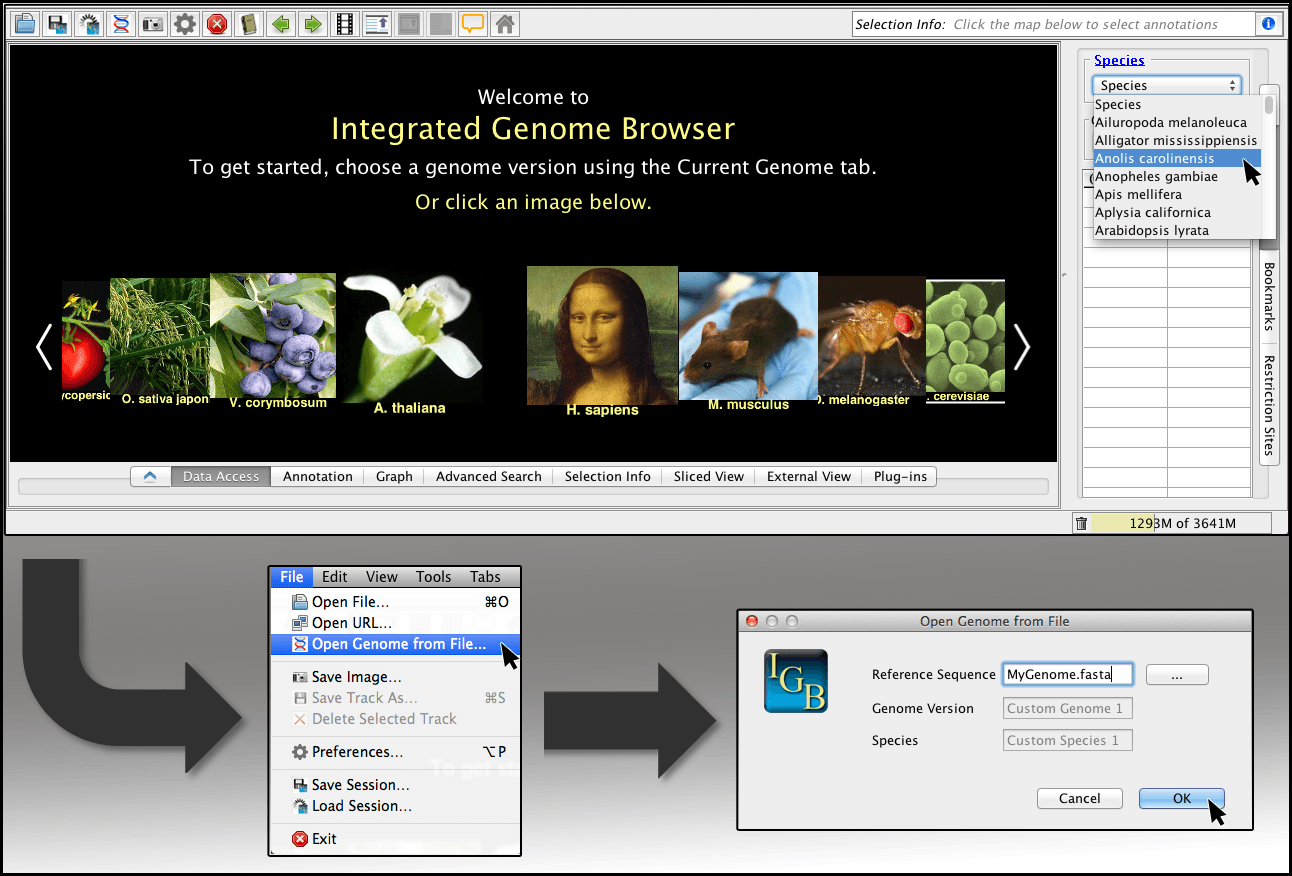
Caractéristiques principales
- Rationalisez votre flux de travail en utilisant des scripts pour définir un génome. Vous pouvez également utiliser R pour gérer l’IGB.
- Créez, personnalisez et enregistrez des graphiques à partir de vos données. Utilisez des graphiques de profondeur pour montrer la couverture ou des graphiques de discordance pour compter les différences entre vos données et une référence.
- Vous pouvez également créer vos propres plug-ins/applications et les partager avec la communauté de l’IGB.
- Vous pouvez créer un QuickLoad IGB pour partager des données entre collaborateurs. Stockez vos données dans le nuage pour permettre à toutes les personnes impliquées dans le projet d’y accéder.
Integrated Genome Browser est un outil utile pour les experts en données, les scientifiques et les bioinformaticiens afin d’automatiser le flux de travail.
DRAGEN
L’analyse DRAGEN trouve des applications dans divers domaines des sciences biologiques, offrant une approche transformatrice de l’analyse génomique. L’analyse DRAGEN permet un examen secondaire précis, approfondi et rationalisé des données de séquençage de la prochaine génération.
L’analyse DRAGEN, qui s’appuie sur la technologie FPGA hautement reconfigurable, accélère les algorithmes d’analyse génomique tout en relevant les défis liés aux temps de calcul et aux volumes de données.
Caractéristiques principales
- Analyse des génomes, exomes, méthylomes et transcriptomes complets à l’aide d’une plateforme unifiée.
- Utilise le génome de référence graphique et l’apprentissage automatique, ce qui permet d’obtenir une précision sans précédent.
- Identifiez et profilez les maladies infectieuses à l’aide d’une solution globale.
- L’analyse DRAGEN offre la flexibilité d’insérer une variété de fichiers d’entrée et de produire une gamme de documents de sortie.
L’analyse DRAGEN s’avère très utile pour l’analyse génomique. Elle est utile aux biologistes computationnels et aux analystes génomiques.
PathAI
PathAI s’appuie sur la vision par ordinateur et l’apprentissage profond pour analyser les images de pathologie, transformant ainsi l’approche traditionnelle du diagnostic et du traitement du cancer.
Les trois principales applications des produits de PathAI consistent à aider les pathologistes à poser de meilleurs diagnostics, à améliorer le développement de médicaments grâce à une utilisation thérapeutique éclairée et à étendre les services de pathologie de premier ordre aux régions qui n’y ont pas accès.

Caractéristiques principales
- Automatise les tâches monotones des pathologistes, améliorant ainsi l’efficacité de l’analyse des lames de pathologie.
- Intègre de manière transparente l’histopathologie aux données génomiques, fournissant des informations précieuses pour éclairer les décisions thérapeutiques.
- Utilise une variété de données et de résultats, ainsi que plusieurs scanners, colorants et sources de laboratoire, afin d’améliorer les prédictions tout en évitant les biais.
En combinant l’expertise des pathologistes avec le logiciel de PathAI, la plateforme libère du temps et permet de se concentrer sur la détermination des options de traitement optimales pour chaque patient.
Geneious
Le logiciel Geneious, développé en Java Swing, présente un haut niveau d’interopérabilité avec les systèmes d’exploitation les plus courants. Il s’agit d’un outil d’analyse biologique complet doté d’une interface conviviale.
Il prend en charge de multiples fonctionnalités telles que les alignements multiples, les arbres phylogénétiques, les assemblages de contigs, les graphiques statistiques, les structures 3D, les chromatogrammes et les électrophérogrammes.

Caractéristiques principales
- Assemblage et analyse de séquençage de nouvelle génération
- Visualisations et graphiques
- Geneious permet de récupérer des bases de données biologiques à partir de différentes plateformes.
- Édition et assemblage de chromatogrammes
- Alignement et phylogénétique
Geneious est un logiciel de bioinformatique très connu, utile pour les chercheurs, les bioinformaticiens et les biologistes computationnels.
MEGA
MEGA, abréviation de Molecular Evolutionary Genetics Analysis, est un logiciel complet conçu pour l’exploration et la visualisation des données de séquences moléculaires. Son objectif principal est d’étudier les relations évolutives intégrées dans les séquences d’ADN ou de protéines.
MEGA fournit aux chercheurs des outils polyvalents qui leur permettent de réaliser des analyses phylogénétiques, d’estimer les distances évolutives et de construire des arbres phylogénétiques détaillés.
Caractéristiques principales
- MEGA permet de construire des arbres phylogénétiques pour visualiser et analyser les relations évolutives entre les séquences biologiques.
- Différentes approches statistiques sont intégrées dans MEGA pour évaluer la signification des résultats des analyses de l’évolution moléculaire.
- MEGA fournit des outils de visualisation pour explorer et interpréter les données moléculaires complexes, y compris les alignements de séquences et les arbres phylogénétiques.
Les chercheurs, les bioinformaticiens et les scientifiques utilisent MEGA pour mieux comprendre l’histoire de l’évolution des gènes, des protéines et d’autres entités biologiques.
BLAST
Le Basic Local Alignment Search Tool (BLAST) identifie les similitudes locales entre les séquences en comparant les séquences de nucléotides ou de protéines à des bases de données. Il calcule la signification statistique des correspondances, ce qui permet de déduire les relations fonctionnelles et évolutives entre les séquences et de faciliter l’identification des membres d’une famille de gènes.
BLAST est un programme et un algorithme bioinformatique largement utilisé pour comparer les informations de séquences biologiques primaires, telles que les séquences d’acides aminés des protéines ou les nucléotides des séquences d’ADN.
Caractéristiques principales
- BLAST est principalement utilisé pour comparer une séquence d’interrogation à une base de données de séquences afin de trouver des séquences similaires ou homologues.
- Il existe plusieurs versions de BLAST, notamment BLAST nucléotide (pour les séquences de nucléotides), BLAST protéine (pour les séquences de protéines), BLASTx (traduit les séquences de nucléotides en séquences de protéines avant de les comparer), et d’autres encore.
- BLAST peut effectuer des recherches dans diverses bases de données biologiques, notamment les bases de données de nucléotides et de protéines du NCBI.
BLAST est un outil essentiel en bioinformatique et en biologie moléculaire, qui permet d’identifier rapidement et précisément les similitudes entre les séquences d’ADN, d’ARN et de protéines.
Clara Parabricks
Clara Parabricks est une suite logicielle rentable pour l’analyse secondaire rapide des données d’ADN et d’ARN NGS, offrant des résultats exceptionnellement rapides par rapport à d’autres méthodes. En tant qu’outil complet d’analyse génomique, Parabricks améliore considérablement le temps de traitement pour des tâches telles que l’analyse de la lignée germinale et de la lignée somatique.
Par exemple, il peut analyser 30x les données du génome humain entier en seulement 25 minutes, ce qui représente une amélioration remarquable par rapport aux 30 heures habituellement nécessaires. Son résultat s’aligne sur les logiciels les plus répandus, ce qui facilite la vérification des résultats.
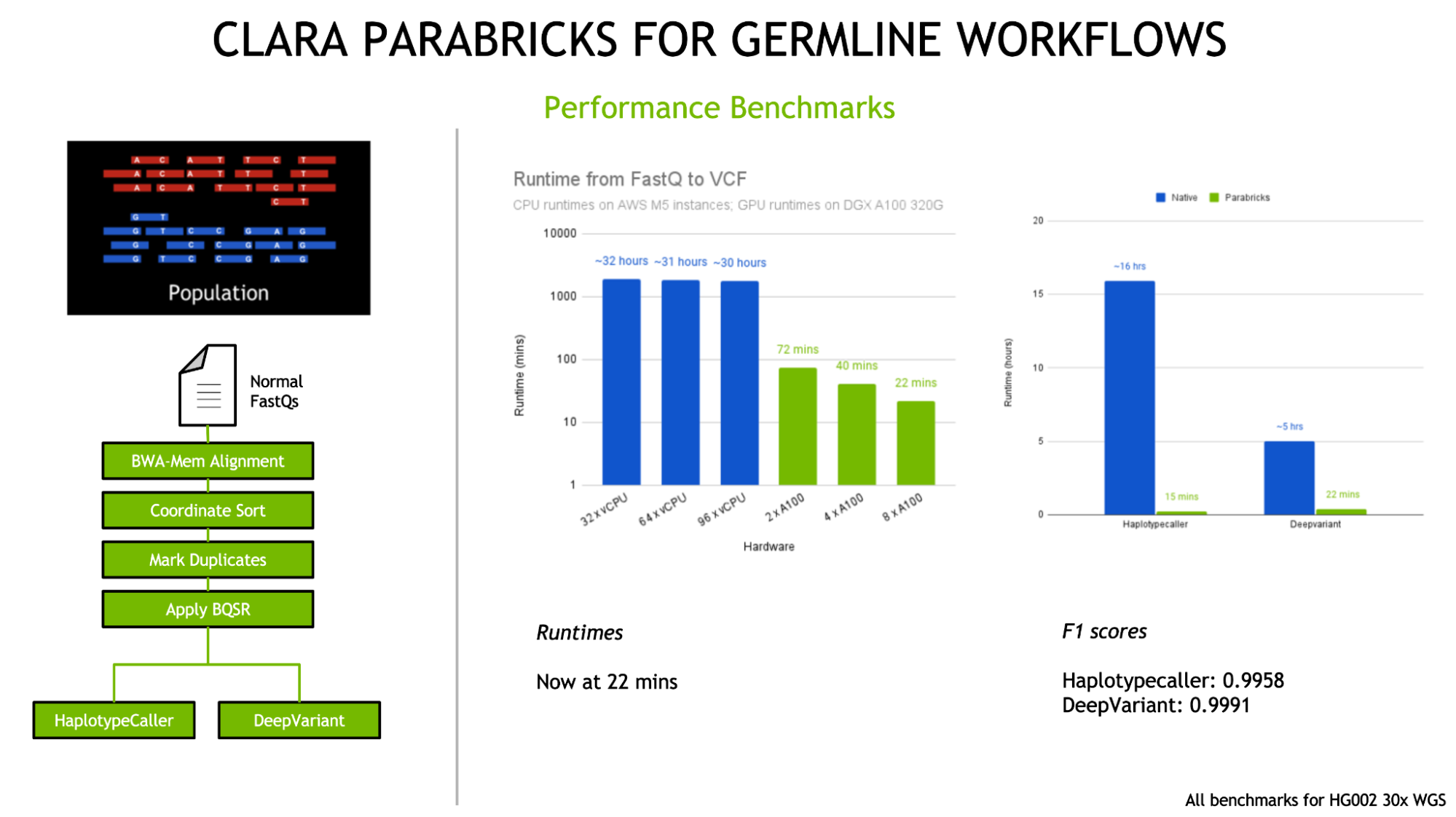
Caractéristiques principales
- Clara Parabricks est uniquement accéléré par le GPU, optimisant l’analyse secondaire avec une vitesse remarquable
- Intégration d’outils basés sur l’apprentissage profond aux côtés des outils standard de l’industrie.
- Réduction significative des temps de calcul et des coûts d’analyse.
- Compatibilité avec Azure et disponibilité gratuite sur NGC.
- Polyvalence dans la prise en charge de divers flux de travail génomiques.
Clara Parabricks est idéal pour les chercheurs en génomique, les bioinformaticiens, les laboratoires cliniques, les entreprises pharmaceutiques, les établissements d’enseignement et les professionnels de la santé.
Pluto
Pluto est une plateforme polyvalente et conviviale qui révolutionne la recherche scientifique. Elle simplifie la gestion des projets, permettant aux équipes d’organiser, de contrôler et d’assigner des expériences avec un partage sécurisé des données pour une collaboration efficace.
Les scientifiques bénéficient de ses outils robustes d’analyse et de visualisation des données, supportant les algorithmes de biologie computationnelle pour une meilleure compréhension de l’expression des gènes, de la liaison ADN-protéine et des biomarqueurs.
Caractéristiques principales
- Permettre aux scientifiques d’effectuer de puissantes analyses bioinformatiques directement dans le navigateur.
- Transformation des données brutes en parcelles prêtes à être publiées en quelques minutes.
- Plate-forme élégante pour l’organisation, le suivi et l’attribution des expériences.
- Outils intuitifs pour l’analyse et la visualisation de données biologiques complexes.
- Compatibilité avec divers essais biologiques, y compris RNA-seq, ChIP-seq, CUT&RUN et essais sur plaques.
Pluto est une plateforme polyvalente conçue pour les utilisateurs de la recherche biologique, de la biologie computationnelle, de la gestion de projets et des efforts scientifiques collaboratifs. Grâce à son interface conviviale et à ses fonctions complètes, elle est utile pour divers rôles au sein de la communauté de la recherche scientifique.
LatchBio
Latchbio est la plateforme la plus flexible pour accéder aux données et les analyser dans le cadre de la R&D biologique. Il s’agit d’un nuage biologique où les organisations peuvent stocker, traiter, analyser et visualiser des données multi-omiques.
Vous pouvez télécharger des flux de travail bioinformatiques dans n’importe quel langage à l’aide du SDK de Latch, recevoir des interfaces associées sans code pour vos scientifiques et bénéficier d’une infrastructure hautement évolutive qui prend en charge l’ensemble de votre entreprise.

Caractéristiques principales
- Latch Registry est une base de données conviviale avec une interface de feuille de calcul conçue pour faciliter la saisie de métadonnées complexes pour les fichiers de séquençage de la prochaine génération (NGS) et les fichiers multi-omiques au sein de la plateforme Latch.
- Latch Data est un système de stockage de fichiers polyvalent capable d’héberger un nombre illimité de données et de fournir un accès universel à tous les membres de votre organisation par le biais d’une connexion unifiée.
- Les Latch Pods se distinguent en tant qu’unités de cloud computing agiles et robustes, avec RStudio et JupyterLab préinstallés pour une analyse en aval sans effort des résultats du flux de travail.
Latchbio vous permet d’accéder à vos données et de les analyser grâce à une interface conviviale. Il s’agit donc d’un outil utile pour les bioinformaticiens, les chercheurs en laboratoire et les chefs d’équipe.
Conclusion
Les outils bioinformatiques sont essentiels pour comprendre et démêler les complexités des données biologiques. De l’organisation des informations à la prédiction des structures protéiques et à l’analyse génomique, ces outils profitent aux chercheurs, aux scientifiques et aux étudiants.
À mesure que la technologie s’améliore, le travail d’équipe entre la biologie et l’informatique nous permet d’en apprendre toujours plus sur la vie. Les outils mentionnés dans cet article vous aideront à analyser les données biologiques, à effectuer des analyses statistiques, à visualiser les données, à appeler des variants, à prédire les structures des protéines, à utiliser les navigateurs génomiques, à effectuer des alignements de séquences, et bien d’autres choses encore.